麻继丹 张锰 王剑 李奋 傅立军 高伟 刘廷亮 张玉奇 沈捷
近年来随着分子遗传学的发展,心肌病致病基因研究成为热点。编码肌小节相关蛋白(粗肌丝和细肌丝组成部分)的基因,如β-肌球蛋白重链(MYH7)基因、肌球蛋白结合蛋白C(MYBPC3)基因、肌钙蛋白T2(TNNT2)基因、肌联蛋白(TTN)基因等[1-2],是常见的心肌病基因。
MYH7基因共40 个外显子,其中38 个外显子编码1 935 个氨基酸[3],构成β-肌球蛋白重链,该蛋白具有3 个功能域,分别为球状头部、头尾接合部(颈部)和杆状尾部(见图1)。在肥厚型心肌病(HCM)有基因突变的患者中,MYH7基因突变患者的比例约为33%[4];
在扩张型心肌病(DCM)有基因突变的患者中,MYH7基因突变患者的比例为4%~8%[5]。目前,已在HCM 和DCM 患者中分别检出186 个和73 个不同位点的MYH7基因突变[6],这些突变位点可能会因编码不同的功能域而产生不同影响[3]。本研究探讨有MYH7基因突变的HCM 或DCM 患儿的临床及基因突变特征,及同一基因突变引起不同类型心肌病的机制。
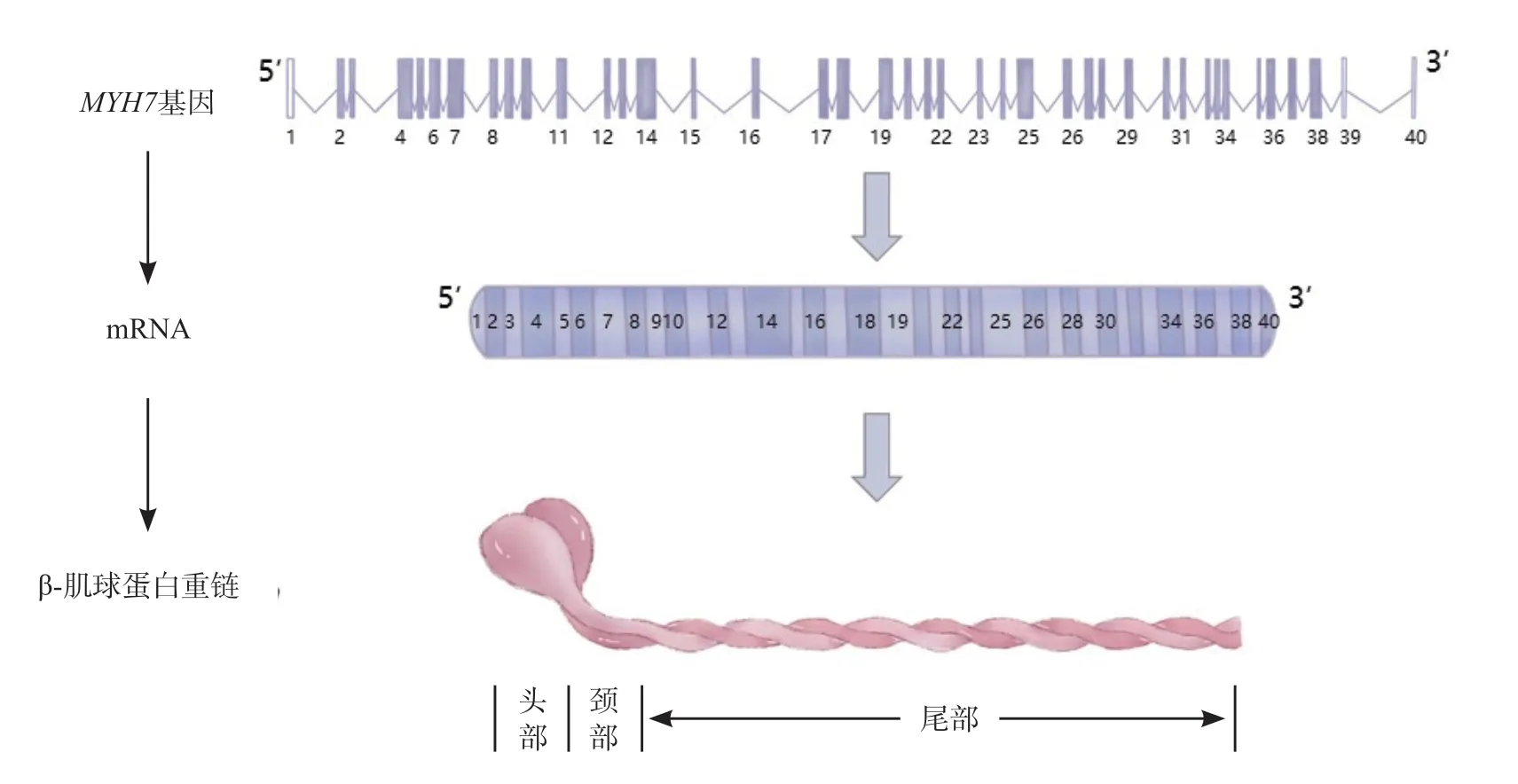
图1 MYH7基因、mRNA、肌球蛋白结构示意图
1.1 研究对象
回顾性收集2015 年1 月至2021 年1 月,在上海儿童医学中心通过基因检测发现的30 例有MYH7基因突变的HCM 和DCM 患儿,设为HCM组和DCM 组。
纳入标准:(1)年龄<18 岁;
(2)通过基因检测发现MYH7基因突变;
(3)HCM 纳入诊断标准为左室后壁舒张期厚度超过正常平均值+2 标准差(按体表面积)或局限性心室壁肥厚,伴或不伴血流动力学流出道梗阻[7]。(4)DCM 纳入标准为左室舒张末期内径(LVEDD)增大超过正常平均值+2 标准差(按体表面积),伴有左室收缩功能减低,如左室射血分数(LVEF)或左室缩短分数(LVFS)减低超过正常平均值-2 标准差(按年龄),或LVEF<45%,LVFS<25%[7]。
排除标准:除外复杂先天性心脏病、高血压性心脏病、缺血性心脏病、心脏瓣膜病变、感染性心肌炎、风湿性心脏病、心包疾病、系统性及遗传代谢性心肌病等患儿。本研究经上海儿童医学中心伦理委员会批准(伦理号:SCMCIRB-K2022045-1)。
1.2 临床资料与基因突变资料收集
收集30 例患儿的临床资料,包括发病年龄、家族史、实验室指标、心电图、超声心动图。收集患儿基因报告,通过NCBI、HGNC、UCSC 等基因数据库或文献检索基因突变位点的报道情况,明确突变位点所在外显子、编码蛋白的结构域及氨基酸的改变。
1.3 统计学分析
使用SPSS 26.0 统计学软件进行统计结果分析。正态分布的计量资料以均数±标准差表示,2 组间比较采用t检验,非正态分布的计量资料使用中位数(四分位数间距)表示,2 组间比较采用秩和检验;
计数资料以例数(百分比)表示,2 组间比较采用Chi-square 检验或 Fisher 精确检验。P<0.05 为差异有统计学意义。
2.1 MYH7基因突变患儿临床特征
共纳入30 例有MYH7基因突变的患儿,其中HCM 患儿21 例,DCM 患儿9 例。HCM 组患儿年龄为(4.0±4.9)岁,12 例为男性,38.1%患儿有阳性家族史。DCM 组患儿年龄为(6.1±5.0)岁,2 例为男性,11.1%患儿有阳性家族史。2 组患儿在性别、诊断年龄、阳性家族史、N 末端脑利钠肽前体(NT-proBNP)、心肌肌钙蛋白(cTnI)、肌酸激酶同工酶MB(CK-MB)以及血清钙离子浓度等方面的差异无统计学意义,在ST 段改变、异常Q 波、异常T 波、传导阻滞(BBB)等心电图异常方面的差异也无统计学意义。见表1。

表1 HCM组与DCM组患儿一般资料与临床特征比较
HCM 组与DCM 组患儿超声心动图指标见表2。HCM 组患儿存在心室肌和室间隔肥厚,室间隔厚度(IVS)为(1.9±1.0)cm,左室后壁厚度(PW)为(0.8±0.4)cm,IVS/PW 比值为2.1±0.6。DCM 组患儿心腔扩张明显,左室收缩功能降低,LVEDD 为(4.2±0.9)cm,左室收缩末期内径(LVEDS)为(3.4±0.8)cm,LVEF 为(42.3±10.2)%,LVFS 为(20.8±5.8)%。

表2 HCM组与DCM组患儿超声心动图指标
2.2 MYH7基因突变谱
本研究在30 例心肌病患儿中检出32 个MYH7基因突变位点,共26 种不同突变位点,分布在17 个外显子中,见表3。24 例(80%)患儿为单位点突变;
6 例(20%)患儿携带多位点突变,包括2 例携带MYH7双突变,4 例携带1 个MYH7突变和其他肌小节基因突变。在32 个突变位点中,除了1 个无义突变c.664C>T(p.Gln222*)、1 个剪切位点突变c.2680-2A>G 及1 个缺失突变c.2539_2541del(p.Lys847del)外,其余29 个均为错义突变。

表3 MYH7基因突变携带者的突变位点
在HCM 组中,20 例(95.2%)患儿基因突变位于肌球蛋白头颈部,1 例(4.8%)患儿基因突变位于肌球蛋白尾部。17 例(81.0%)患儿基因突变来源于父亲或母亲,3 例(14.3%)患儿基因突变来源于新生突变,1 例(4.7%)患儿基因突变来源不详(母亲未留样)。
在DCM 组中,8 例(88.9%)患儿基因突变位于肌球蛋白头颈部,1 例(11.1%)患儿基因突变位于肌球蛋白尾部。6 例(66.7%)患儿基因突变来源于新生突变,2 例(11.1%)患儿基因突变来源于父亲或母亲,1 例(11.1%)患儿基因突变来源不详(母亲未留样)。
本研究发现的26 个突变位点中,16 例为已报道致病性突变,10 例为国内外未曾报道的致病性突变,其中3 例为HCM,7 例为DCM。见表3。
3.1 MYH7基因突变与临床表型的关系及机制
MYH7基因编码的β-肌球蛋白重链,是肌小节中介导肌丝滑行的重要组成部分,具有3 个功能域。本研究在30 例患儿中共检出MYH7基因26 种不同突变位点,分布在16 个外显子中,以错义突变为主。
MYH7基因不同位点的突变可导致HCM 和DCM 两种不同临床表型。有多种假说解释其潜在的病理生理机制,主流观点有突变蛋白的剂量效应和钙离子通道敏感性变化等。
3.1.1 突变蛋白的剂量效应 MYH7 突变蛋白的剂量效应与该基因突变引起2 种不同临床表型密切相关。突变蛋白的杂合状态更易导致HCM 表型,而纯合状态更易引起DCM 表型,最终导致不可逆的心力衰竭[6]。既往研究发现,MYH7杂合子突变小鼠更多地表现为HCM 中常见的肥厚表型,纯合子突变小鼠却更接近DCM 表型,会发生进行性心室扩张,造成新生小鼠的死亡[8-9]。
MYH7基因致病性突变大多以常染色体显性遗传的错义突变为主[10]。本研究发现在HCM 患者中,MYH7基因突变患者以错义突变为主,且均为杂合突变,所有患儿无进行性心室扩张表现。Tanjore等[6]在MYH7基因突变患者中发现,同一个位点突变下,杂合突变患者多表现为HCM,纯合突变患者多表现为DCM,表明临床表型可能与突变蛋白的剂量效应相关。
本研究虽然未在DCM 患儿中发现纯合突变,但有2 例DCM 患者存在MYH7基因复合杂合突变。
患者1 为MYH7基因c.427C>T(p.Arg143Trp)(杂合)错义变异,联合c.664C>T(p.Gln222*)(杂合)无义变异,其父亲携带无义变异c.664C>T(p.Gln222*)(杂合),母亲携带错义变异c.427C>T(p.Arg143Trp)(杂合),该患儿发病年龄8 个月,临床出现了为较严重的扩张型心肌病表现,且存在家族猝死史。
患者2 为MYH7基因c.2821C>T(p.Arg941Cys)(杂合)错义变异,合并存在c.2680-2A>G(杂合)剪接位点变异,其中错义变异为新生突变,剪接位点变异为父亲携带。该患儿临床表型为DCM,发病年龄5 个月,且出现较为严重的心功能不全表现。
Hershkovitz 等[11]的研究也发现类似突变,在1个家系中,2例DCM患儿检出MYH7基因c.427C>T(p.Arg143Trp)联 合c.4588C>T(p.Arg1530*)多位点突变,其中1 例患儿(姐姐)1 岁时诊断为DCM,接受了心脏移植手术,但仍于3 岁时死亡,另1 例患儿(弟弟)于3 个月龄时在住院期间死亡。该家系中MYH7基因c.427C>T 单位点突变携带者发病年龄晚,临床表现为较轻的HCM 或无症状。
由此推测,复杂的基因型(复合杂合子或纯合子)通常与早期发病和不良临床表型相关[12]。MYH7基因序列中突变剂量可影响临床表型、发病年龄和疾病严重程度等。MYH7基因突变剂量较高者(复合杂合子或纯合子),临床表现较严重,多以预后较差的DCM 为主。
3.1.2 钙离子通道敏感性变化 钙离子在维持心肌细胞正常收缩节律中起重要作用。心肌细胞内钙离子水平升高,可增强心肌细胞收缩力;
钙离子内流减少,则心肌收缩力减弱[13]。目前认为,心肌细胞膜对钙离子敏感性改变可影响心肌细胞收缩力,从而导致MYH7单基因突变出现2 种截然不同的临床表型。
心肌细胞膜对钙离子敏感性增加可导致心肌高收缩性,从而促进肥厚表型[14-15]。而血浆中游离钙离子水平增加,会进一步损伤心肌组织的舒张功能。Montag 等[16]研究证实,MYH7基因突变的HCM 患者合并有心肌细胞钙离子敏感性的变化,且心肌细胞钙离子通道敏感性存在显著的异质性,可能与MYH7突变位点不同有关,也可能与具有相同MYH7基因突变的患儿对钙离子抑制剂的反应性不同有关。
心肌细胞膜对钙离子敏感性降低可导致心肌收缩力下降,引起心脏收缩功能障碍,从而表现为DCM 表型[17]。同时,肌小梁钙离子通道敏感性受损和细胞骨架蛋白的应力变化,也会进一步导致心室扩张的DCM 表型。
本研究并未发现MYH7突变患儿的血钙离子水平异常,目前仍需进一步证实钙离子通道敏感性的变化与MYH7基因变异位点的关系,为钙脱敏剂和钙致敏剂等HCM 和DCM 靶向用药提供理论支撑。
3.1.3 表观遗传修饰 随着研究深入,有学者认为心肌病同一致病基因突变引起疾病表型的差异,不仅与DNA 编码序列改变有关,而且与基因表达的修饰相关。表观遗传学是独立于DNA 序列变化调节基因表达的机制,主要通过DNA 甲基化、组蛋白修饰、染色质重塑、非编码RNA 等修饰作用,调节染色质结构和转录因子结合,进而调节基因表达。表观遗传修饰及其上游信号的急性变化与生理、病理刺激及饮食、压力、体育活动等环境因素有关[18]。
3.2 新发现突变位点
本研究发现10 个国内外未曾报道的MYH7突变位点,其中3 个位点表现为HCM,7 个位点表现为DCM。曾有报道分析了MYH7 的蛋白质结构,模拟计算了可能出现的致病性突变位点,其中计算机模拟位点c.2494C>T(p.Leu832Phe)突变有致病可能[19],但尚无研究报道,本研究首次报道且证实携带该突变位点的为HCM 患者。
本研究在1 例DCM 患儿中检测出MYH7基因c.2821C>T(p.Arg941Cys)位点突变,该突变在既往HCM 或DCM 患者研究中未报道,但曾报道过在1 例左室心肌致密化不全患者中检出[20],这进一步体现了心肌病的异质性。
MYH7基因其他位点如c.664C>G(p.Gln222*)、c.790G>C(p.Glu264Gln)、c.1103A>G(p.Gln368Arg)、c.1607A>G(p.Glu536Gly)、c.2295C>G(p.Phe765Leu)、c.2663A>C(p.Gln888Pro)、c.2617C>G(p.Leu873Val)、c.2680-2A>G 等突变,尚无文献报道在正常人群或心肌病人群中检出,本研究首次在HCM 或DCM 患者中检出。
3.3 小结
本研究为单中心回顾性研究,存在以下局限性:(1)样本量偏小,可能存在选择偏倚。(2)部分患儿发病年龄较小,表述不清,症状不明显,检查配合程度受限,可能存在临床资料偏差。(3)部分轻症或外地患儿未能规律随访,缺少长期随访资料。(4)心肌病的发病机制包括基因突变和环境因素,需要进一步实验证明基因及环境因素对心肌病表型的共同影响。
不同位点的MYH7基因突变可引起HCM 或DCM 两种完全不同的临床表型。同一基因突变可引起不同临床表型,说明了心肌病的遗传异质性,其机制尚不明确,可能与表观遗传学修饰相关。不同心肌病在儿童期或疾病早期临床表现可无明显差异,分子遗传学检测可帮助疾病的早期识别、预后判断、生育咨询以及发病机制研究。
猜你喜欢错义杂合心肌病不明原因新生儿高胆红素血症与UGT1A1基因突变的关系*广西医科大学学报(2022年5期)2022-06-07GJA8基因错义突变致先天性白内障一家系遗传分析昆明医科大学学报(2022年3期)2022-04-19甘蓝型油菜隐性上位互作核不育系统不育系材料选育中常见的育性分离及基因型判断种子(2021年3期)2021-04-12MKRN3基因与儿童中枢性性早熟中南医学科学杂志(2019年6期)2019-12-05伴有心肌MRI延迟强化的应激性心肌病1例中国临床医学影像杂志(2019年1期)2019-04-25扩张型心肌病中医辨治体会中国中医药信息杂志(2016年5期)2016-12-01从翻译到文化杂合——“译创”理论的虚涵数意外语教学理论与实践(2016年1期)2016-06-11TAKO-TSUBO心肌病研究进展国际心血管病杂志(2015年5期)2015-02-27CYP2A13基因8种错义突变与肺癌易感性的关联研究浙江医学(2014年17期)2014-04-13雄激素可调节的肾脏近端肾小管上皮细胞靶向杂合启动子的优化华东理工大学学报(自然科学版)(2014年1期)2014-02-27