杨春晓, 张艳琨, 杨 蒙, 彭国建, 张可欣, 郭丽芳, 夏福婷
(云南民族大学 云南省高校民族地区资源清洁转化重点实验室, 昆明650500)
作为一种非常规气态污染物,AsH3是含砷化合物中毒性最高的物质,对环境和人体健康均具有巨大的危害. 它还是制约黄磷尾气、电石炉尾气、典型密闭冶金炉窑尾气中利用高浓度CO生产化工产品的关键阻碍之一[1-8]. 化学吸收法、吸附法、燃烧法等是目前已报到的AsH3的主要净化方法[9]. 但是这些方法存在易造成腐蚀设备、难以全量捕集氧化砷和造成二次污染等问题[10-15]. 近年来,催化氧化法被广泛应用于AsH3的去除,它利用金属氧化物、氧气和砷的协同作用使之生成砷或砷氧化物等固体物质,可借助催化剂表面固定氧化砷产物,不仅提高了AsH3的去除效率,而且可减少二次污染[16],符合绿色环保的理念. 因此催化氧化法是公认的净化矿冶尾气中AsH3的最佳方法[17, 18].
在AsH3的净化过程中,实验风险和实验成本均很高,而且实验条件苛刻. 因此目前国内外对AsH3污染消除的实验研究并不多见. Yilong Lin[19]等人通过制备不同合成条件下的类水滑石化合物(HTLC)前体,表明AsH3的去除率与CuZnAl类水滑石催化剂的合成条件密切相关. Cu-Fe和Cu-Mn二元类水滑石催化剂也体现出较强的AsH3去除性能[20]. 此外经Cu(NO3)2改性的Hp型分子筛对AsH3有更好的吸附性能[21]. 在外加磁场作用下,Fe/HZSM-5催化剂能有效脱除AsH3是因为催化剂表面生成了更多的Fe2O3活性位点,促进了AsH3的催化氧化[22]. 与实验研究相比,砷污染消除的理论研究越来越多[23-30]. 计算研究表明掺杂了过渡金属的碳纳米管(SWCNTs)对AsH3气体分子具有更好的吸附性能[27]. 由于Be、Mg、Ca和Sr的强化学反应性,碱土元素掺杂的石墨烯对AsH3分子表现出更强的化学吸附,促进了碱土掺杂石墨烯在气体分子检测领域的应用[28]. 此外,具有强吸附能力和优异稳定性的单层Hf2CO2在室温下可作为AsH3气体吸附剂的高效候选材料,尤其在外加电场时,可以通过偶极极化有效提高AsH3与单层Hf2CO2相互作用强度,实现AsH3的可逆吸收[29]. 在无催化剂时,用密度泛函理论(DFT)研究了水对AsH3的氧化过程的影响,发现存在水时反应势垒能降低3.27~10.70 kcal/mol,表明水对AsH3的氧化过程有促进催化作用[30].
目前对AsH3气体分子的理论模拟多以吸附为主,对催化反应机理的研究相对较少. 值得关注的是,砷与铁氧化物或铁氢氧化物具有高度亲和性,铁氧化物因为其表面积大、表面电荷高等特点对许多化学层面的污染物都表现出较强的吸附能力,可以有效的去除砷,不同铁氧化物的组成和结晶形态的不同导致对砷的吸附能力也有所区别[31-35]. 因此研究AsH3在铁氧化物上氧化的机理至关重要. 有研究者结合实验和第一性原理计算研究了小的中性氧化铁簇(FeO1-3和Fe2O4,5)与一氧化碳(CO)的反应,通过FemOn团簇与CO的反应为理解CO与冷凝相催化剂活性位点相互作用的内在机理提供一个很好的模型[36]. 本文拟采用密度泛函理论探究Fe2O3上AsH3的催化氧化反应机理,以期为AsH3催化氧化高效催化剂的研制起指导作用.
B3LYP方法因计算准确性和耗时合理性而被广泛运用于含有过渡金属体系的计算中[37,38]. 本文采用密度泛函理论(DFT)[39-41]在Gaussian03程序包[42]中对反应体系中所有的反应复合物(RC)、过渡态(TS)、中间体(INT)和产物复合物(PC)使用B3LYP杂化泛函进行计算,对于Fe原子,采用包含相对论修正的LANL2DZ赝势基组,对于As、H和O原子使用6-31++G(d, p)基组. 以分离的反应物的能量之和作为零点,对各驻点的频率进行考察,全实频的是稳定点,有唯一虚频的是过渡态. 通过内禀反应坐标(IRC)[43, 44]计算,以确定与过渡态相关联的反应物以及产物,为了获得更加精确的能量,我们对总能量的零点能(ZPE)[45]、焓和吉布斯自由能在温度为298.15 K、压力为1.0 atm时进行校正. 本文研究了Fe2O3上AsH3的催化氧化反应机理. 以Fe2O3作为催化剂来进行铁氧化物上AsH3催化氧化反应机理的理论计算探究,以Fe2O3中的两个Fe原子为不同的活性中心进行研究,每个活性中心均设计了3个步骤. 反应途径如下:
图1为AsH3、Fe2O3和O2在B3LYP/6-31++G(d, p)水平下优化的几何构型. AsH3中As-H的键长为1.457 Å;
Fe2O3中Fe1-O1、Fe1-O2和Fe1-O3的键长分别为1.773 Å、1.761 Å和1.771 Å;
Fe2-O1、Fe2-O2和Fe2-O3的键长分别为1.771 Å、1.763 Å和1.772 Å.

图1 AsH3、Fe2O3和O2在B3LYP/6-31++G(d, p)水平下几何构型Fig. 1 Geometric configurations of AsH3, Fe2O3 and O2 at the level of B3LYP/6-31++G(d, p)
为了对两个路径的反应研究得到更多的信息,本文计算了催化剂Fe2O3和反应复合物RC1、RC2的Mulliken电荷密度,如图2所示. 在Fe2O3单体中,两个Fe原子所处的化学环境一致,所带电荷相同. RC1的活性中心为Fe1,Fe1所带电荷为1.123 e,O1和O2是与Fe1直接相连的两个O原子,所带电荷分别为-0.630 e和-0.304 e. 同理,RC2则表示活性中心为Fe2,Fe2、O1和O2所带电荷分别为1.127 e、-0.585 e和-0.564 e. 由此可知Fe2原子所带正电荷更大以及O1、O2的负电荷之和也大于RC1. 因此在RC2中Fe原子与O原子之间的静电吸引更强. 此外,RC1和RC2的吸附能分别为103.72 kcal/mol和51.60 kcal/mol,从吸附能也可以看出RC2更稳定. 所以推测反应开始,由于AsH3和O2的吸附,会让Fe2O3发生形变.

图2 Fe2O3、RC1和RC2在B3LYP/6-31++G(d, p)水平下几何构型Fig. 2 Geometric configurations of Fe2O3, RC1 and RC2 at the level of B3LYP/6-31++G(d, p)
3.1 活性中心为Fe1
如图3所示反应的第一步是从反应复合物RC1到中间体INT1-1,对应着Fe2O3催化剂上一分子AsH3与一分子O2反应. 在RC1到INT1-1中,过渡态TS1-1的唯一虚频为-1035.62 icm-1,其振动模式对应着O2原子进攻As原子的同时,H1原子转移到O1原子上. 在TS1中,As-O2的原子距离为1.761 Å,比RC1中该键长长了0.008 Å. 翻越了TS1这个过渡态,得到中间体INT1-1,从过渡态TS1-1到中间体INT1-1的过程中,As原子和H1原子之间的距离从1.636 Å伸长至2.701 Å;
H1原子和O1原子之间的距离从1.636 Å缩短至0.983 Å,表明第一步进行的是As-H1键的断裂和H1-O1键的形成.

图3 Fe2O3(活性中心Fe1)催化剂上(AsH3+O2)在气相中各驻点在B3LYP/6-31++G(d, p)水平下优化得到的几何构型图,键长单位为Å;
相对自由能的单位为kcal/molFig.3 The stagnation points of (AsH3+O2) on Fe2O3 (active center Fe1) catalyst in the gas phase are at the level of B3LYP/6-31++G(D, P), the unit of bond length is Å; the unit of relative free energy is kcal/mol

图4 Fe2O3(活性中心为Fe1)催化剂上(AsH3+2O2)在气相中各驻点在B3LYP/6-31++G(d, p)水平下优化得到的几何构型图,键长单位为Å;
相对自由能的单位为kcal/molFig. 4 The optimized geometrical configuration diagrams of (AsH3+2O2) on Fe2O3 (active center is Fe1) catalyst in the gas phase at the level of B3LYP/6-31++G(d, p), the unit of bond length is Å; the unit of relative free energy is kcal/mol
在第二步INT1-2到INT1-3中,是INT1-1+O2经过构型优化得到了中间体INT1-2(图4). 由于新加的O2分子对H1原子有吸引,已经转移到O1原子上的H1原子会暂时转移到O4原子上. 在中间体INT1-2之后,出现了过渡态TS1-2,对其振动频率分析可知,TS1-2在反应历程中只有一个虚频,值为-871.64 icm-1. 从过渡态的振动模式可以看出O4原子进攻As原子的同时,H2原子转移到O3原子上. 在INT1-2中,As-H2的键长为1.517 Å,而在TS1-2和INT1-3中,该键长变为1.539 Å和3.233 Å,伸长了接近一倍,与此同时,H2-O3原子间距从TS1-2中的1.801 Å 缩短至INT1-3中的0.971 Å,说明此时As-H2键的完全断裂以及H2-O3键的形成.
如图5所示,在INT1-3上又加入一分子O2经过构型优化得到了中间体INT1-4. TS1-3的唯一虚频为-817.60 icm-1,振动模式对应着O6原子进攻As原子的同时,H3原子转移到O5原子上. 从INT1-4到产物复合物PC1中,As-H3原子之间的距离分别为1.490 Å、1.559 Å 和3.169 Å ,增长了将近1.679 Å. 而从TS1-3到PC1的过程中,H3-O5的键长从1.800 Å缩短至0.971 Å,表明在得到产物复合物PC1的过程中,主要发生的是As-H3键的断裂和H3-O5键的形成.

图5 Fe2O3(活性中心为Fe1)催化剂上(AsH3+3O2)在气相中各驻点在B3LYP/6-31++G(d, p)水平下优化得到的几何构型图,键长单位为Å;
相对自由能的单位为kcal/molFig. 5 The optimized geometrical configuration diagrams of (AsH3+3O2) on Fe2O3 (active center is the Fe1) catalyst in the gas phase at the level of B3LYP/6-31++G(d, p), the unit of bond length is Å; the unit of relative free energy is kcal/mol

图6 Fe2O3(活性中心为Fe2)催化剂上(AsH3+O2)在气相中各驻点在B3LYP/6-31++G(d, p)水平下优化得到的几何构型图,键长单位为Å;
相对自由能的单位为kcal/molFig. 6 The geometric configuration diagrams obtained by optimizing the stagnation points of Fe2O3 (active center is the Fe2) catalyst (AsH3+O2) in the gas phase at the level of B3LYP/6-31++G(d, p), the unit of bond length is Å; the unit of relative free energy is kcal/mol
3.2 活性中心为Fe2
当活性中心为Fe2时,AsH3的氧化同样分三步完成. 首先由一分子的AsH3和一分子O2经过优化得到反应复合物RC2. 当AsH3分子中的H1原子靠近O2分子中的O1原子时,O2原子转移到AsH3分子中As原子上,从而得到了过渡态TS2-1. 从RC2到第一个中间体INT2-1中,其间经历一个具有唯一虚频且虚频值为-938.57 icm-1的过渡态TS2-1. 如图6所示,过渡态TS2-1的结构特征为O2原子进攻As原子,同时伴随着值O2原子转移到AsH3分子中As原子上. 在TS2-1中,As-H1的键长为1.550 Å,H1-O1的键长为1.632 Å,O1-O2的键长为2.567 Å,As-O2的键长为1.730 Å. 反应经过渡态之后,As-O2的键长增加0.111 Å;
As-H1的键长增加1.346 Å;
O1-O2的键长增加0.043 Å;
H1-O1的键长减少0.658 Å,形成第一个中间体INT2-1.
接下来的一步如图7所示,由INT2-1+O2经过构型优化得到中间体INT2-2,然后出现了一个连接INT2-2和INT2-3的过渡态TS2-2,其虚频值是-986.35 icm-1. 对此虚频的振动模式分析表明此过渡态对应着As-H2键的伸长和H2-O3键的缩短. 如图所示,从TS2-2中As-H2原子之间的距离为1.550 Å,比INT2-3短了将近1.465 Å,而TS2-2的H2-O3键长比INT2-3长了将近0.744 Å. 这些变化表明得到中间体INT2-3的过程中主要进行的是As-H2键的断裂和H2-O3键的形成.

图7 Fe2O3(活性中心为Fe2)催化剂上(AsH3+2O2)在气相中各驻点在B3LYP/6-31++G(d, p)水平下优化得到的几何构型图,键长单位为Å;
相对自由能的单位为kcal/molFig. 7 The geometric configurations of the catalyst (AsH3+2O2) on the Fe2O3 (active center is the Fe2) catalyst in the gas phase at the B3LYP/6-31++G(d, p) level,the unit of bond length is Å; the unit of relative free energy is kcal/mol
在得到最终产物复合物PC2的最后一步中,又加入了1分子O2,AsH3分子中的H3原子靠近O2分子中的O5原子时,O6原子转移到AsH3分子中的As原子上,得到了过渡态TS2-3(对应的唯一虚频为-838.69 icm-1). 如图8所示,过渡态TS2-3的结构特征为O6原子进攻As原子,同时伴随着H3原子转移到O5原子上. H3-O5键的形成可以从TS2-3中的键长1.827 Å缩短至PC2中的0.972 Å 看出. 离去键As-H3键长从TS2-2中的1.541 Å变为PC2中的3.136 Å,表明As-H3键的逐渐断裂以及最终产物复合物PC2的形成.
表1为B3LYP/6-31++G(d, p)水平下AsH3和O2在Fe2O3上进行催化氧化反应的反应复合物、中间体复合物、产物复合物各驻点的能量数据,单位是kcal/mol. 其中△Ee表示电子能量,△E0表示零点振动能,△ETRV=△Ee+转动能,△H表示焓变,△S表示熵变,△G表示吉布斯自由能,△G≠表示自由能垒.
活性中心为1号铁原子的反应物RC1的吉布斯自由能△G为103.72 kcal/mol、过渡态TS1的△G为117.37 kcal/mol,INT1的△G为101.91kcal/mol,第一步所对应的活化吉布斯自由能垒△G≠为13.65 kcal/mol. 第二步中,INT1-2、TS1-2和INT1-3的△G分别为56.01 kcal/mol、106.00 kcal/mol和46.88 kcal/mol,第二步所对应的活化吉布斯自由能垒△G≠为49.99 kcal/mol,第三步中INT1-4、TS1-3和产物PC1的△G为38.41 kcal/mol、53.05kcal/mol和-9.77 kcal/mol,第三步所对应的活化吉布斯自由能垒△G≠为14.64 kcal/mol. 由此可见,第二步的自由能垒△G≠为49.99 kcal/mol比第一步的自由能垒△G≠为13.65 kcal/mol和第三步的自由能垒△G≠为14.64 kcal/mol要高,由此可见第二步是该过程的速度控制步骤.
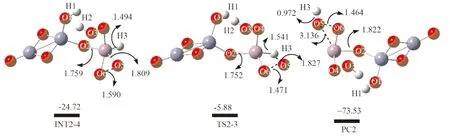
图8 Fe2O3(活性中心为Fe2)催化剂上(AsH3+3O2))在气相中各驻点在B3LYP/6-31++G(d, p)水平下优化得到的几何构型图,键长单位为Å;
相对自由能的单位为kcal/molFig. 8 The geometric configuration diagrams obtained by optimizing the stagnation points of Fe2O3 (active center is the Fe2) catalyst (AsH3+3O2) in the gas phase at the level of B3LYP/6-31++G(d, p), the unit of bond length is Å; the unit of relative free energy is kcal/mol
当活性中心为2号铁原子时,也进行了三步反应. 第一步中,反应物复合物RC2的吉布斯自由能△G为51.60 kcal/mol、过渡态TS2-1的△G为66.09 kcal/mol,INT2-1的△G为27.58 kcal/mol,因此第一步所对应的活化吉布斯自由能垒△G≠为14.49 kcal/mol,后面两步的活化吉布斯自由能垒△G≠分别为21.20 kcal/mol和18.84 kcal/mol. 由此可见第二步的△G≠最高,即该步骤为速度控制步骤. 比较铁原子的两种活性中心可知,活性中心为2号铁原子的速度控制步骤的自由能垒较低,故在Fe2O3上AsH3催化氧化中活性中心为2号铁原子附近反应更容易发生. 此外,之前的研究表明,在无催化剂存在下,AsH3直接氧化生成砷氧四面体的过程中速率控制步骤的能垒为50.14 kcal/mol. 而通过本文研究发现,活性中心为1号铁原子时的能垒为49.99 kcal/mol,与无催化剂时的直接氧化相比能垒几乎不变. 但活性中心为2号铁原子时的能垒(21.20 kcal/mol)却大大降低. 所以推测反应开始,由于AsH3和O2的吸附,会让Fe2O3发生形变. 通过比较自由能垒可知,反应更倾向于发生在正电荷丰富的区域.

表1 B3LYP/6-31++G(d, p)水平下Fe2O3上AsH3催化氧化反应各驻点在气相中相对于分离反应物的能量(单位为kcal/mol)
本文采用密度泛函理论研究了Fe2O3上AsH3催化氧化的反应机理,研究考虑了Fe2O3中的两个Fe原子为不同的活性中心. 通过反应过程中各驻点的能量、键长以及分子振动状态以及电荷分析可知,活性中心为2号铁原子的速度控制步骤的自由能垒较低,故在Fe2O3上AsH3催化氧化中活性中心为2号铁原子附近反应更容易发生,比直接氧化(50.14 kcal/mol)低28.94 kcal/mol. 此研究将为AsH3催化氧化反应高效催化剂的寻找和和开发奠定理论基础.
猜你喜欢中间体驻点复合物碳量子点黄芩素复合物对金黄色葡萄球菌抑菌作用的研究九江学院学报(自然科学版)(2022年2期)2022-07-02建立A注射液中间体中肉桂酸含量测定方法中国药学药品知识仓库(2022年10期)2022-05-29三氧化二砷三元复合物纳米递送系统的构建及评价中草药(2022年5期)2022-03-03线粒体泛醌氧化是肿瘤生长的必要条件实用肿瘤学杂志(2021年3期)2021-11-29氟代六元氮杂环化合物与水分子弱相互作用的理论研究西华大学学报(自然科学版)(2021年3期)2021-05-17药品稳定性在药品质量控制中的运用研究山东工业技术(2018年9期)2018-05-26利用远教站点,落实驻点干部带学党的生活·党员电教与远程教育(2016年3期)2016-02-26随县教育局与帮扶户“结对认亲”湖北教育·综合资讯(2016年1期)2016-02-192300名干部进村“串户”办实事源流(2015年8期)2015-09-16原料药:抗生素中间体价格上涨股市动态分析(2015年12期)2015-09-10